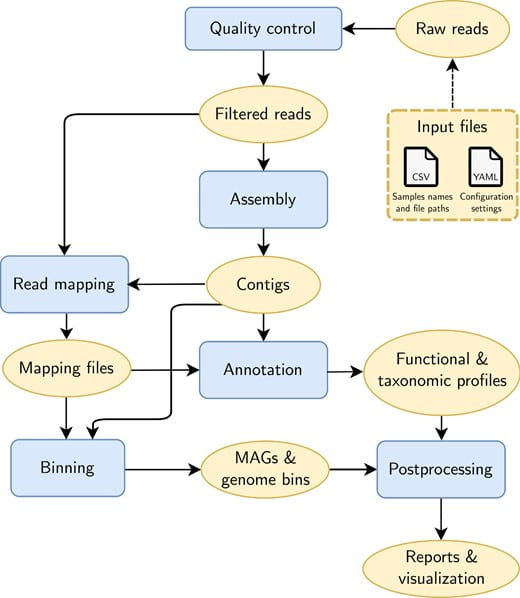
Metaphor: A workflow for streamlined assembly and binning of metagenomes
Genome-resolved metagenomics techniques aim to recover genomes from high-throughput sequencing data, and have led to unprecedented insight into microbial diversity, ecology, and evolution.
However, data pipelines for for assembling and binning metagenomes are inherently complex, have high computing cost, use heterogeneous data sources, have dozens of customizable parameters, and depend on several specialized bioinformatics software.
PhD student Vinícius Salazar introduces Metaphor, building on the work of late Bobbie Shaban, MetaGenePipe at Melbourne Integrative Genomics, and in collaboration with MDAP.
Metaphor is an automated and flexible workflow for the assembly and binning of metagenomes. It recovers prokaryotic genomes from metagenomes efficiently and with high sensitivity, and it provides taxonomic and functional abundance data for quantitative metagenome analyses. Metaphor can process multiple datasets in a single execution. Abundance data can be easily imported into downstream analysis tools. Detailed performance metrics provide users with a high-level summary of their analysis.
Vinícius W Salazar, , Metaphor—A workflow for streamlined assembly and binning of metagenomes, GigaScience, Volume 12, 2023, giad055, https://doi.org/10.1093/gigascience/giad055
Project homepage: https://github.com/vinisalazar/metaphor
Documentation: https://metaphor-workflow.readthedocs.io/
Operating system(s): Linux, Mac OS (Intel)
Programming language: Snakemake (Python 3)
Other requirements: Conda, Snakemake v7 or higher, Python 3.7 or higher.
Categories